

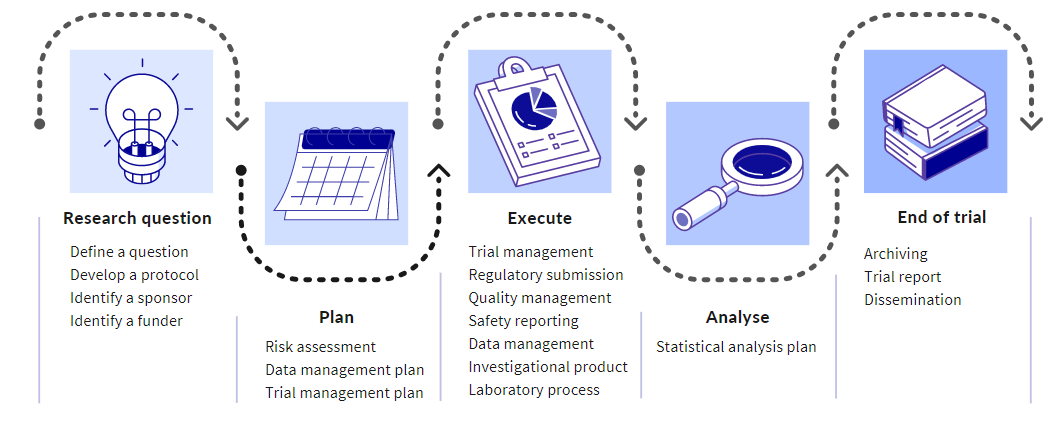
Rare Diseases Clinical Trials Toolbox




Would you like to learn more about the toolbox and its use? View the short instructional video here.
Read also our publication explaining how the tool was developed and the methodology used: "Rare diseases clinical trials toolbox - public resources and main considerations to set up a clinical trial on medicinal products for humans in Europe": here.
If you have a tool that you think should be added to the toolbox please complete and return the application form.
You can download the full list of tools here.
Latest update: 02/12/2025
DEFINE A QUESTION
The definition of the research question is key to research design. All research must have a primary question, clearly stated in advance, and founded on a systematic review of what is already known. Researchers who plan studies without reviewing what has been done, risk performing research for which the answer is already known or exposing participants to ineffective or an inferior treatment.
Evidence suggests that research protocols often lack important information on study design. The study protocol should provide an adequate explanation for why the proposed study methodology is appropriate for the question posed, why the study design is likely to answer the research question, and why it is the best approach.
The planning of a clinical trial depends on the primary question, and researchers should clearly and simply explain in the study protocol what the trial is aiming to show, why it is worth asking and, through consultation with public and patient groups, why this is worthwhile to patients. The primary question should be consistently stated throughout the study protocol.
The question should generally include specific information on participants, intervention(s), comparator(s) and outcomes (PICO format):
- Population: what are the characteristics of the patient or population-e.g. condition?
- Intervention: what is the intervention under consideration for this patient or population-e.g. a drug or surgical intervention?
- Comparison: what is the alternative to the intervention-e.g. a different drug or a placebo?
- Outcomes: what are the key outcomes the study would measure and how do they answer the primary question? e.g. mortality, disease progression, surrogate parameter?
| Tool Name | Relevance | Year | RD/ Paediatric specific | Type |
| Clinical Trial Decision Tool | Clinical trial decision tool developed by Paul Janssen Futurelab and the CCMO. The tool helps you in assessing whether your study is considered a clinical trial, a low-intervention clinical trial or is outside scope according to the definitions given in the Clinical Trials Regulation. | 2022 | No | Guideline |
| Cochrane PICO search | PICO searchBETA allows you to use PICO terms to find the Cochrane Reviews most relevant to your healthcare question. In particular, it allows you to find reviews in which a term is used specifically as a population, an intervention, a comparison, or an outcome | 2020 | No | Search tool |
| Patient group engagement Priotization Tool | Web-based prioritization tool to help clinical research sponsors and patient groups identify high-priority engagement activities. Use of this tool can help sponsors and patient groups identify the engagement activities that they believe will provide the most benefit for the least investment | 2020 | No | Toolbox |
| Patient engagement resource centre | Selected relevant public resources to help researchers understand the basics of patient engagement, and guide you through the different phases of patient engagement: from planning to conducting and evaluating. | 2024 | No | Toolbox |
| Short guide on patient partnership in rare disease research projects | The European Joint Programme on Rare Diseases (EJP RD) has developed, together with a working group led by EURORDIS, named Patient Engagement in Biomedical Research Projects (PENREP), a short guide on Patient Partnership in rare disease research projects. | 2022 | Yes | Guideline |
| EUPATI patient involvement resource library | EUPATI Patient Involvement Resource Library, is a peer reviewed collection covering patient involvement in medicines development, including advocacy, regulation, clinical trials and digital health. The resources are free to use and share with proper referencing. | 2025 | No | Toolbox |
DEVELOP A PROTOCOL
The ICH GCP E6 (R3) (International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use- Good Clinical Practice) guideline defines the protocol as “A document that describes the objective(s), design, methodology, statistical considerations and organisation of a trial. The protocol usually also gives the background and rationale for the trial, but these could be provided in other protocol-referenced documents”
The research protocol explains how the proposed study methodology is appropriated for the question posed, demonstrate that the design is likely to answer the research question and why it is the best approach. The protocol should explain:
- the rational, background information and literature review
- how the relevant successes and failures of previous studies have been taken into account in the design of the planned trial
- how the proposed research method is appropriate for the question posed
- the reasoning behind the choice of any treatment/intervention difference sought, as well as the other parameters used in the determination of the sample size
- the choice of comparators and end points
- the randomisation and blinding methods
- the suitability of the statistical tests
- how the sample to be studied is representative to the wider group of patients
The SPIRIT (Standard Protocol Items: Recommendations for Interventional Trials) statement 2025 is a guideline for the minimum content of a clinical trial protocol. It includes a 34-items checklist that applies to protocols for all clinical trials and focuses on the content on a clinical trial protocol.
| Tool Name | Relevance | Year | RD/ Paediatric specific? | Type |
| ICH General considerations for clinical studies E8 (R1) | Describes internationally accepted principles and practices in the conduct of both individual clinical trials and overall development strategy for new medical products | 2021 | No | Guideline |
| ICH General principles for planning and design of multi-regional clinical trials E17 | Describes general principals for the planning and design of randomised multinational clinical trials with the aim of increasing its acceptability in global regulatory submissions | 2017 | No | Guideline |
| ICH Clinical investigation of medicinal products in the paediatric population E11 (R1) | Provides an outline of critical issues in pediatric drug development and approaches to the safe, efficient, and ethical study of medicinal products in the pediatric population. The purpose of this addendum is to complement, provide clarification and current regulatory perspective on topics in pediatric drug development. | 2017 | Yes | Guideline |
| Preparedness of medicines’ clinical trials in paediatrics | Developed by the European Network of Paediatric Research (Enpr-EMA) working group, these recommendations are a structured assessment of contributing factors that enable efficient conduct of individual clinical trials in paediatrics and were developed for sponsors, principal investigators and triallists. | 2020 | Yes | Recommendation |
| ICH Harmonised guideline paediatric extrapolation E11A | This guideline provides recommendations for harmonised approaches for paediatric extrapolation to support the development and authorisation of paediatric medicines. This guideline is to be read in conjunction with the ICH E11 guideline on clinical investigation of medicinal products in the paediatric population | 2024 | Yes | Guideline |
| European Medicines Agency Guidance for Applicants seeking scientific advice and protocol assistance | This guidance document addresses a number of questions that users of the scientific advice or protocol assistance procedures may have. It provides an overview of the procedure to obtain scientific advice or protocol assistance and gives guidance to Applicants in preparing their request. This guidance document also explains the scope and nature of scientific advice and protocol assistance. It will enable Applicants to submit requests which are in line with Scientific Advice Working Party (SAWP) requirements and which can be validated and evaluated quickly and efficiently. | Regular updates | No | Guideline |
| Advice on medicines for human use in the EU medicines regulatory network | "With a view to clarifying the scope of current scientific and regulatory advice activities, ACT-EU Priority Action on consolidated advice has mapped information on current voluntary procedures available from EU regulators on Medicines for Human use and collated this information in the form of questions and answers" | 2024 | No | Guideline |
| IRDiRC Building block_ Alternative designs for small population clinical trials | General recommendations to select the most efficient study design for each medical condition or trial and on potential adaptations of conventional designs to the low sample size scenario | 2020 | Yes | Guideline |
| EMA Guideline on clinical trials in small populations | This Guideline considers problems associated with clinical trials when there are limited numbers of patients available to study. It has been prepared by the CHMP (Committee for Medicinal Products for Human Use) Efficacy Working Party in joint collaboration with members of the Scientific Advice Working Party (SAWP), the Committee on Orphan Medicinal Products (COMP) and the Paediatric Expert Group (PEG). | 2007 | Yes | Guideline |
| Design and analysis of clinical trials for small rare disease populations (Hilgers et al., 2016) | This paper refers to the current state of design and analysis methods, as well as practical conditions to be considered when conducting a clinical trial for rare diseases. | 2016 | Yes | Article |
| Recommendations for the design of small population clinical trials (Day et al., 2018) | Recommendations of the IRDiRC expert group on clinical trials for RD around six topics: different study methods/designs and their relation to different characteristics of medical conditions, adequate safety data, multi-arm trial designs, decision analytic approaches and rational approaches to adjusting levels of evidence, extrapolation, and patients’ engagement in study design | 2018 | Yes | Article/Guideline |
| Clinical trial designs for rare diseases: Studies developed and discussed by the International Rare Cancers Initiative (Bogaerts et al., 2015) | The IRCI (International Rare Cancers Initiative) trials are each presented to exemplify possible approaches to designing credible trials in rare cancers. Researchers may consider these for use in future trials and understand the choices made for each design. | 2015 | Yes | Article |
| Opinions and letters of support on the qualification of novel methodologies for medicine development | The European Medicines Agency (EMA) publishes opinions on the qualification of innovative development methods and letters of support for novel methodologies that have been shown to be promising in the context of research and development into pharmaceuticals. | Regular updates | No | Recommendation |
| COMET: Core outcome measures in effectiveness trials | The COMET Initiative brings together people interested in the development and application of agreed standardised sets of outcomes, known as ‘core outcome sets’ (COS). These sets represent the minimum that should be measured and reported in all clinical trials of a specific condition, but COS are also suitable for use in routine care, clinical audit and research other than randomised trials | Regular updates | No | Checklist |
| Cochrane Central Register of controlled trials | The Cochrane Central Register of Controlled Trials (CENTRAL) is a highly concentrated source of reports of randomized and quasi-randomized controlled trials. In addition to bibliographic details (author, source, year, etc.) CENTRAL records will often include an abstract (a summary of the article). They do not contain the full text of the article. | Regular updates | No | Registry |
| 2025 SPIRIT Statement (Standard Protocol items) | Reporting guideline defining standard protocol items for clinical trials. The evidence-based SPIRIT recommendations were developed using systematic, transparent methodology and broad consultation with 115 experts representing diverse stakeholders involved in the design, funding, conduct, review, and publication of trial protocols. | 2025 | No | Checklist |
| Guidelines for Reporting Outcomes in Trial Protocols The SPIRIT-Outcomes 2022 Extension | This SPIRIT-Outcomes 2022 extension of the SPIRIT 2013 statement provides 9 outcome-specific items that should be addressed in all trial protocols and may help increase trial utility, replicability, and transparency and may minimize the risk of selective nonreporting of trial results. | 2022 | No | Checklist |
| SPIRIT Statement (Standard Protocol items) for n-of-1 trials | Extension to the SPIRIT (standard protocol items: recommendations for interventional trials) guideline, SPENT (SPIRIT extension for n-of-1 trials), to improve the completeness and transparency of n-of-1 trial protocols. | 2019 | Yes | Checklist |
| SPIRIT PRO Extension for inclusion of patient-reported outcomes in clinical trials protocols | Extension of the SPIRIT (Standard protocol items: recommendations for interventional trials) guideline, SPIRIT PRO provides guidelines for inclusion of patient-reported outcomes in clinical trial protocols. | 2018 | No | Checklist |
| Measuring health-related quality of life in patients with rare disease (Lenderking et al., 2021) | This article explores some of the challenges in HRQoL assessment in rare disease, propose solutions, and consider regulatory issues | 2021 | Yes | Article |
| Patient reported outcome measures in rare diseases: a narrative review (Slade et al., 2018) | This review explores some of the current issues around the utilisation of PROMs in rare diseases, including small patient populations and dearth of valid PROMs. Difficulties in validating new or current PROMs for use in clinical trials and research are discussed | 2018 | Yes | Article |
| PROMs Repository | The ERICA Patient Reported Outcome Measures (PROMs) Repository is the first attempt to identify and centralize Clinical Assessment Outcomes questionnaires of relevance for rare diseases and constitutes a milestone in the Europe-wide standardization of Patient-Centered Outcome Measures (PCOMs) and PROMs for rare diseases. | Regular updates | Yes | Repository |
| COSMIN: Database of systematic reviews of outcome measurement instruments | Database of systematic reviews of outcome measurement instruments | Regular updates | No | Database |
| PARADIGM patient engagement toolbox | This project deliverable centralises all PARADIGM’s co-created recommendations, tools and relevant background information to make patient engagement in medicines development easier for all. The toolbox could help develop clinical trials with a further enhanced patient-focus and improve the experience of patients participating in the trials. Developed by PARADIGM project. | 2020 | No | Toolbox |
| EUPATIConnect | EUPATIConnect matches patient experts with researchers to create mutually beneficial opportunities and to enhance the future of patient engagement. | 2022 | No | Advisory board |
| EURORDIS Community Advisory Board (CAB) Programme | Patient Community Advisory Boards (CABs) are groups established and operated by patient advocates. They offer their expertise to sponsors of clinical research. For example, by being involved before a clinical study starts, patients help ensure that clinical studies are designed to take into account their real needs, resulting in higher quality research. | NA | Yes | Advisory Board |
| European YPAG Network | The network was established to support the development of new Young Person Advisory Groups (YPAGs) within Europe. The main aim of eYPAGnet is to provide researchers with a variety of opportunities to work with children and young people in the design and conduct of paediatric clinical trials. Here are just some of the services we offer. | NA | Yes | Advisory Board |
| EDCTP Protocol development tool | An initiative of the Global Health Network, this Protocol Development Toolkit has been developed to support researchers in this process, to provide the tools and guidance to produce a high-quality health research Protocol. | Regular updates | No | Toolbox |
| Assessment of short-term outcome of neonatal trials: Points to consider | PedCRIN tool: This tool lists examples of data items for the assessment of short term efficacy and safety outcomes of neonatal trials | 2021 | Yes | Guideline |
| Assessment of long-term outcome of neonatal trials: Points to consider | PedCRIN tool: This tool lists examples of data items for the assessment of long-term efficacy and safety outcomes of neonatal trials | 2021 | Yes | Guideline |
| Protocol development for neonatal trials: Points to consider for pharmacovigilance | PedCRIN tool: This tool gives points to consider concerning pharmacovigilance and risk management at the time neonatal protocol development | 2021 | Yes | Guideline |
| Exclusion criteria in neonatal trial protocols: Points to consider | PedCRIN tool: This tool gives points to consider concerning pharmacovigilance and risk management at the time neonatal protocol development | 2021 | Yes | Guideline |
IDENTIFY A SPONSOR
The ICH for Good Clinical Practice guidelines E6 (R3) and the Clinical Trials Regulation (536/2014), define a sponsor as
“an individual, company, institution or organisation which takes responsibility for the initiation, for the management and for setting up the financing of the clinical trial”
According to this definition, the sponsor is equally scientifically, legally and financially responsible of a clinical trial but the budget can either come from the sponsor itself or from sources external to the research group.
The sponsor is ultimately responsible for the scientific, ethical, regulatory and legal aspects of the trial, and also for financial aspects (i.e., if an external funder withdraws, the sponsor will be responsible for looking for funds to complete the trial).
The latest European regulation for clinical trials on medicinal products for human use (536/2014) introduces the possibility of co-sponsorship in Europe, while for Sponsors located out of the EU a legal representative of the sponsor is needed.
Industry-sponsored trials
In commercial research, the same organization (usually a pharmaceutical company), funds, designs and carries out a trial, acting both as sponsor and funder.
Investigator-initiated trial (IITs)/Academic-sponsored trials
Commercially sponsored clinical trials are responsible for bringing most of the new drugs to the market. However, these clinical trials only assess the safety and efficacy of drugs that are chosen by a commercial entity that funds the entire process. Non-commercial or academic trials have therefore their own additional specific significance, often focusing on refining indications of available treatments and optimizing therapeutic strategies that do not have as much financial gain to the pharmaceutical industry.
A good amount of IITs is driven by questions that generally arise beyond the completion of Phase III studies and which have not been studied during Phases I–III of drug development.
For IITs, academic institutions act usually as sponsor (academic-sponsored trials), but not necessarily as the funder. Only in few cases will academic sponsors be able to conduct trials without external funding, usually coming from different sources. This often leads to complex arrangements with multiple partners joining in international consortia. The setup of a complex multi-national partnership may sometimes be at odds with the concept of “single sponsorship” which was developed for ensuring protection of participants in the context of commercial trials. Single sponsorship is rooted in the need to clearly identify the legal responsibility and does not hinge upon the funding aspect. Noteworthy, most funding agencies are unwilling to take the role of sponsor and often they may not be suitable for it.
Sponsor-Investigator is an individual who both initiates and conducts, alone or with others, a clinical trial, and under whose immediate direction the investigational product is administered to, dispensed to, or used by a subject. The term does not include any person other than an individual (e.g., it does not include a corporation or an agency). The obligations of a sponsor-investigator include both those of a sponsor and those of an investigator.
Collaboration with industry - For some IITs, pharmaceutical companies might act as funders. The pharmaceutical company can support IITs with drug supply, funding, material and/or information, as allowed under local laws and regulations, provided that they align with the company defined areas of strategic interest. Some big pharma companies publish guidelines describing the conditions to establish collaborations with academic-sponsors for IITs.
| Tool Name | Relevance | Year | RD/Paediatric specific | Type |
| None identified |
FIND A FUNDER
Industry-initiated clinical trials are financially supported by the industry. The principal investigator (PI) salary and the costs associated with running the trial are all covered by the pharmaceutical company that conceived the clinical trial. In investigator-initiated trials (IIT), however, usually is the PI who applies for funding through research programs and government grants to fund their conceived research project.
Industry funded trials
Industry- funded and industry-sponsored clinical research is paid by an industry organization that has contracted with a faculty member to conduct a clinical trial that involves an intervention with, or observation of, a disease or biomedical condition, or a registry/repository related to a disease or biomedical condition. For most industry-funded clinical trials, the industry organization acts as sponsor and designs the study, owns the protocol, data and results.
Academic-sponsored trials funded by industry
Collaboration between industry and academics is common in the development of vaccines, drugs, and devices, as it can be mutually beneficial. Academic institutions/hospitals provide access to trial participants and clinical and methodological expertise, and industry provides funding and expertise. The degree of independence and the roles of academics and industry vary across trials. Trials may be run by academic trial units independently but with unrestricted industry funding, or the only contribution from industry could be free provision of study drugs.
Publicly funded trials
Many national research funding agencies open regularly calls for funding clinical research, supporting academia/investigators to set up clinical trials. This funding is nevertheless usually restricted at national level. Indeed, despite the advantages of multinational clinical trials, just 3% of academic trials (vs. 30% of industry trials) involve more than one country. In Europe, the relative scarcity of multinational academic trials can be explained, in part, by restrictions with current cross-border funding options.
Funding by a central European Budget
Horizon 2020 (H2020) has been the biggest EU Research and Innovation programme ever, with nearly €80 billion of funding available over seven years (2014 to 2020). Most multinational clinical trials have been funded under the H2020 “Health, demographic change and wellbeing” programme. Since 2021, the Program Horizon Europe (HE) is the new Framework Program which will run from 2021 to 2027. With an indicative budget or around €95.5 billion it is expected that upcoming calls will include funding for clinical studies and clinical trials for rare diseases.
European and industry funding combined
The Innovative Health Initiative (IHI) is a Horizon Europe source of funding dedicated to public-private partnership projects. Non-industry partners in consortia receive public funding, while industry contributions can be in cash (financial contributions) and/or in-kind (such as personnel time, laboratory access, data, compounds) depending on the partner. A large share of each project must come from industry contribution (≥ 45%).
Cross-border national funding
In general, national funding agencies do not accept funding crossing borders, limiting international collaboration.
Some national public funding agencies (German DFG, Austrian FWF and Swiss SNSF) have established mechanisms to coordinate national calls for funding, facilitating the creation of “virtual common pots”, through mutual opening of the respective funding procedures that facilitate the implementation of cross-border research projects.
Founded by the Nordic Council of Ministers, NordForsk funds clinical studies, including clinical trials, following both the “virtual and real common pot” models or mixed models combining both. NordForsk can enter into collaboration with funding organisations/institutions outside the Nordic Region when this is expected to result in high scientific quality and Nordic added value. The virtual common pot model is the preferred funding model for partners from outside the Nordics where NordForsk can act as a broker between national funding organisations in the Nordics and organisations outside the Nordics.
Benefit is a funding initiative involving KCE and ZonMW, national funders for Clinical studies in Belgium and Netherlands. The program funds pragmatic Trials running in both countries, following the “virtual common pot” model.
There are also private initiatives supporting multinational trials, pooling funding sources. The GCRFF (Global cardiovascular Research Funders Forum) facilitates simultaneous request of funding to charities located in different countries. The ATTRACT programme launches single calls pooling funding from charities located in different countries under the virtual common pot model with limited cross-border funding.
Co-funded Partnerships
Co-funded partnerships combine national funding from EU member states and European Commission funding. The co-financing rate is typically 30 % from the EC. They offer opportunities for funding through the launch of Joint Transnational Calls. Joint Transnational Calls are based on the “virtual common pot” funding model. Through this mechanism, each national funder supports the components of the applying consortium located in their own country. ERDERA is the current rare-diseases specific co-funded (50 % EC co-fund) partnership. ERDERA started in 2024 and will be running until 2031. The partnership will launch one call for clinical trials.
| Tool Name | Relevance | Year | RD/Paediatric specific | Type |
| Exploring new uses for existing drugs: innovative mechanisms to fund independent clinical research (Verbaanderd et al., 2021) | This paper describes and discusses funding opportunities for independent clinical repurposing research | 2021 | No | Article |
| Scientify research | An open, curated and structured research funding database | Regular updates | No | Database |
| NIRO (Navigating Innovation & Research Opportunities) | This tool is designed to help single entities or teams from the private and public sector, not-for-profit organisations, and academia identify which R&I initiatives and programme opportunities could be the right fit, including initiatives and programmes sponsored by the European Commission, as well as national ministries, agencies and not-for-profit organisations within and beyond the European Union. | Regular updates | No | Inventory |
| Overview of currently open calls within the Cluster Health area (EU) | Overview of currently open calls proposed by HNN. 3.0, a Horizon Europe funded project aiming to align services of national contact points of Horizon Europe Cluster Health. Among others, they are developing tools to make easier finding funding calls on the Cluster Health area. | Regular updates | No | Inventory |
| Database for multicountry clinical research funding opportunities in Europe | Developed by the ERA4Health Partnership, this document is an inventory of funding resources available for multinational investigator-initiated clinical studies in Europe | 2023 | No | Inventory |
RISK ASSESSMENT
Risk assessment is a systematic process for identifying and evaluating events that could affect the achievement of clinical study´s objectives related to quality, safety, timelines and budget, positively or negatively.
Some academic organisations may not be in a position to undertake the role of sponsor for clinical trials or may only sponsor trials of a certain risk level. It is essential therefore that a risk assessment is undertaken at the very start. The process could be defined such that the risk assessment is undertaken on the research proposal and then further refined once the protocol has been drafted.
The risk-based approach relies on the identification and assessment of risk(s) and mitigation of these risks.
For years, risk has been interpreted as risk for patient's safety or rights only. However, other types of risk should be considered: the institutions and teams in charge of the study conduct, the governance structures, the target population, and the public health stakeholders, etc.
The risks of a clinical trial on a medicinal product depend on a number of factors but can be broadly categorised as:
- the risks of the investigational medicinal product (IMP)
- the risks associated with the regulatory and legal requirements, the trial conduct, design and methods
The OECD Recommendation on the governance of Clinical Trials introduced a risk-based oversight and management methodology for clinical trials. It combines a stratified approach that is based on the marketing authorisation status of the medical product and can be applied in a common manner across countries’ regulatory frameworks, with a trial-specific approach that considers other issues such as the type of populations concerned by the trial, or the informed consent of the patients. EMA Reflection paper on risk-based quality management in clinical trials (2013) and EC considerations on Risk Proportionate approaches in clinical trials (2017) aim to facilitate the development of systematic, prioritised, risk-based approach to quality management of clinical trials, to support the principles of Good Clinical Practice and to complement existing quality practices, requirements and standards.
Importantly from a risk perspective, ICH E6 (R3) emphasises a risk-based and proportionate approach to monitoring, oversight, trial conduct. The guideline structure (principles & objectives + Annex 1 for interventional trials + Annex 2 for non-traditional trials) further supports tailoring of risk assessment to trial design.
| Tool Name | Relevance | Year | RD/Paediatric specific | Type |
| OECD Recommendation on governance of clinical trials | To facilitate international co-operation in clinical trials on medicinal products, particularly for trials initiated by academic institutions, in December 2012 the OECD Council adopted a set of principles calling for improved consistency among national regulations and their interpretations, and on streamlined procedures for the oversight and management of clinical trials. This framework introduces a risk-based oversight and management methodology for clinical trials. | 2013 | No | Recommendation |
| Guideline on strategies to identify and mitigate risks for first-in-human and early clinical trials with investigational medicinal products | The guideline is intended to further assist stakeholders in the transition from non-clinical to early clinical development and in identifying factors influencing risk for new investigational medicinal products (IMPs). The document includes considerations on quality aspects, non-clinical and clinical testing strategies, study design and on the conduct of FIH/early CTs | 2017 | No | Guideline |
| Risk assessement form for clinical research projects | Swiss Clinical Trials Organisation Tools: Risk Assessment of potential risks of a clinical research project-in line with current requirements (ICH GCP E6(R2)) | Regular updates | No | Guideline |
TRIAL MANAGEMENT PLAN
The purpose of a Project Management Plan (PMP) in a clinical trial is to define the scope, outline responsibilities and describe key steps of the clinical trial process. This not only ensures that those performing the tasks have a clear plan of what, when and how trial activities are undertaken, but also enables auditors/inspectors to reconstruct how a trial was managed. Trial management procedures/responsibilities that are common across all trials are usually defined in the sponsor’s research standard operating procedures (SOPs)/policies and for less complex trials, the trial management documentation may simply consist of an index of these SOPs with any trial-specific management described in sufficient detail in the protocol. The level of detail required will depend upon:
a) The clinical trial risk assessment
b) The organisational structure within which the clinical trial is conducted
c) The design and methods of the clinical trial
The trial management plan should outline procedures regarding:
- Communication
- Documentation (Trial Master File)
- Vendors
- Investigational product
- Site management
- Safety plan
- Data management (Data Management plan)
| Tool Name | Relevance | Year | RD/Paediatric specific | Type |
| None identified |
DATA MANAGEMENT PLAN (DMP)
DMP is a written document that describes the plans for collection and management of data throughout the lifecycle of a clinical trial. The DMP describes which clinical data will be acquired and how it will be handled, stored, checked for consistency and plausibility, and made available for the final analysis and further research after the end of the project.
For effective data management, planning must begin at the time of trial design. It should consider the collection and management of data during the trial, data sharing and archiving at trial closure. A well-designed DMP will provide a road map on how to handle the data, establish processes to handle unforeseeable conditions and assess potential risks.
As there are various stakeholders and staff members involved in data handling, the key goal of this document is to communicate to each stakeholder the required information to create and maintain a high-quality database that is ready for analysis. In addition to the database specification, DMP should also document CRF (Case report Form) design and development.
It is considered best practice to develop the DMP in collaboration with all stakeholders involved in the trial, to ensure compliance. Institutes and agencies involved in clinical research might consider developing DMP templates, which are customised as per the requirements of the trial.
Typical data management plan elements:
- Data collection-Types of data, contextual details (metadata)
- Data Storage: Storage, backup and security
- Data Protection: Access, monitoring, provisions for protection/privacy
- Policies for re-use
- Access and sharing
- Archiving
Special attention must be given to data access, sharing and re-use. As of July 1, 2018, manuscripts submitted to ICMJE (International Committee of Medical Journal Editors (ICMJE)) journals that report the results of clinical trials must contain a data sharing statement as described below.
Data sharing statements must indicate the following: whether individual deidentified participant data (including data dictionaries) will be shared; what data in particular will be shared; whether additional, related documents will be available (e.g., study protocol, statistical analysis plan, etc.); when the data will become available and for how long; by what access criteria data will be shared (including with whom, for what types of analyses, and by what mechanism)
Because of the scarcity of patients and the need to share information internationally to find similar cases, data sharing and ‘matchmaking’ is imperative for rare disease research. Notably, the challenge in establishing consent policies for rare disease research stems from the dichotomy between the push for free-flow of data against concerns about loss of privacy. Informed consent, access governance, and access technologies are important to realize the benefits of data sharing while mitigating risks and should be taken in consideration while designing a Data Management Plan.
| Tool Name | Relevance | Year | RD/Paediatric specific | Type |
| Guidelines for effective Data Management Plan | Guidance to create Data Management plans developed by the Inter-university Consortium for Political and Social Research (ICPSR), an international consortium of academic institutions and research organizations. | Regular updates | No | Guideline |
| Data Management Plan Online | Templates for data management plans based on the specific requirements listed in funder policy documents. | Regular updates | No | Template |
TRIAL MANAGEMENT
Trial management is the process of ensuring that a trial is run effectively and within budget and timelines. According to ICH, the sponsor should utilize appropriately qualified individuals to supervise the overall conduct of the trial, to handle the data, to verify the data, to conduct the statistical analyses, and to prepare the trial reports. The sponsor should implement a system to manage quality throughout all stages of the trial process
For industry-sponsored trials, the sponsor usually outsources this activity to a CROs-private Clinical Research Organizations. Although there are different types of CROs and diverse levels of specialization (distinct therapeutic areas for instance), typical CRO services include regulatory affairs, site selection and activation, recruitment support, monitoring, data management, trial logistics, pharmacovigilance, biostatistics, medical writing, and project management, among others.
For academic-sponsored trials, Trial Management is usually performed by an academic Trial Coordinating Centre.
The trial Coordinating Centre is at the heart of the trial, whether it is a single-site or multisite trial and is usually linked to the Sponsor ´s institution (Hospital or University).
The Trial Coordination Center can be referred to by a variety of names and set in many different environments. It can be:
- An academic clinical trials unit (CTU) able to provide all the services
- An office/desk in a clinical department in a hospital/university
- Split over different locations and be set up as a combination of the above for example a CTU can support an academic department in one or more aspects of running a trial (data management, statistics etc).
As responsible of the project management, CROs and Trial Coordinating Centers act as Global coordinators of resources, including, but not limited to:
- Clinical site feasibility
- General coordination of resources
- Preparation and maintenance of sponsor’s trial master file
- Organisation of pathways for central laboratory/biobank with instructions for single sites
- Elaboration of the monitoring manual
- Central clinical research associate (CRA) training (web-based and/or on-site)
- Online, web-based central monitoring
- Review of monitoring reports
The sponsor may transfer any or all of the sponsor's trial-related duties and functions to a CRO/Trial Coordinating Center but the ultimate responsibility for the quality and integrity of the trial data always resides with the sponsor. The CRO/TCC should implement quality assurance and quality control
| Tool Name | Relevance | Year | RD/ Paediatric specific | Type |
| Mapping of organisations providing support to multicountry IICS | This mapping describes organizations that have developed capacity for supporting planning and designing multicountry Investigator Initiated Clinical Studies | 2023 | No | Inventory |
| Guidance for good randomized trials (The Good Clinical Trials Collaborative) | The guidance is intended to support all individuals and organizations involved in the planning, conduct, analysis, oversight, interpretation, funding, and regulation of RCTs of any health intervention for any purpose in any setting. | Regular updates | No | Guideline |
| Cambridge Clinical trials Unit SOPs and Documents | Cambridge Clinical Trials Units SOPs and templates on: pre-trial planning, protocol development, set-up; pharmacovigilance; data management and statistics; sample management; trial management; post-study procedures and archiving | Regular updates | No | Template |
| Global Health Trials Tools and Templates library | Developed by the Global Health Trials Knowledge Hub , this library of templates includes clinical trials´ general logs and trackers, documents for finances management, patients enrolment and study, site and staff management | Regular updates | No | Template |
| CTTI implementation tools | CTTI-developed tools to improve the quality and efficiency of clinical trials. Tools include resources to optimize recruitment and informed consent process | Regular updates | No | Toolbox |
| UKTMN Guide to Efficient Trial Management | This guideline describes the process of managing clinical trials and gives an overview of the trial management framework, both legal and operational, providing hints, tips and references to external resources | 2024 | No | Guideline |
| NCCIH Clinical Research Toolbox | This toolbox contains templates, sample forms, and information materials to assist clinical investigators in the development and conduct of high-quality clinical research studies | Regular updates | No | Toolbox |
| PORTICO Clinical Trials Toolkit | The toolkit is a curated series of publicly accessible videos and links covering essential operational details that early investigators need to know before embarking on a clinical trial with a special focus on pediatric trials. Topics address study startup, study documents, consent, oversight, management, and safety. | Annual updates | Yes | Toolbox |
| PANDA: A practical Adaptive & Novel Designs and Analysis toolkit | PANDA is aimed at trialists and researchers in clinical trials who are keen to learn about adaptive designs, their practical application, potential benefits and limitations. The target audience includes, but is not limited to, trial statisticians, clinicians, health economists, grant proposal developers, trial managers, data managers, and reviewers of grant applications | Regular Updates | No | Toolbox |
| The adaptative platform trial toolbox | This toolbox aims to collect the accumulated knowledge, experience, & resources from multiple projects and trials into a practical and guided toolbox to facilitate planning & conduct of future APTs in any therapeutic area. | 2023 | No | Toolbox |
| EnprEMA Network Database | This database includes research networks and centres with recognised expertise in performing clinical studies in children. It is part of the European network of paediatric research at the European Medicines Agency (Enpr-EMA). | Regular updates | Yes | Database |
| Feasibility assessment of neonatal studies and selection of investigator sites/ study centres: Points to consider | PedCRIN tool: This tool lists examples of points to consider for the feasibility assessment and selection of neonatal centres | 2021 | Yes | Recommendation |
| Improving inclusion of under-served groups in clinical research: Guidance from INCLUDE project | This guidance summarises what an under-served group is, a roadmap suggesting intervention points to improve inclusion, examples of under-served groups and barriers to inclusion | 2020 | Yes | Guidance |
| Enrolment into neonatal trials: Points to consider during protocol development | PedCRIN tool: This tool provides a list of points to consider during protocol development for improving the enrolment of neonatal trials | 2021 | Yes | Recommendation |
| Ethical considerations for clinical trials on medicinal products conducted with minors | Recommendations of the expert group on clinical trials for the implementation of Regulation (EU) No 536/2014 on clinical trials on medicinal products for human use | 2017 | Yes | Recommendation |
| Informed Consent for Paediatric Trials in Europe | This document lists the country-specific requirements for informed consents for paediatric clinical trials in Europe | 2015 | Yes | Guideline |
| Assent / Informed Consent Guidance for Paediatric Clinical Trials with Medicinal Products in Europe | Developed by Enpr-EMA’s Working Group on Ethics, this document is intended to be used as an overview tool of the contents for assent/informed consent forms for all stakeholders (such as patients, sponsors and investigators) to support the conduct of high quality paediatric clinical trials in Europe across all paediatric age groups, from birth to less than 18 years of age. | 2021 | Yes | Guideline |
| Library of participant information leaflets | This repository of Participation Information Leaflets (PILs) and Informed Consent Forms (ICFs) used in randomised trials was collected as part of the EXCELSIOR project. The repository of PIL/ICFs will allow researchers to review the different ways in which trial concepts are described and use these to develop their own future participant information leaflets and consent forms. | 2020 | No | Inventory |
| ICF template | ICF template model developed by the IMI funded Do-it project. It means to cover all the information to comply with GDPR rules | 2019 | No | Template |
| ICF guidelines | ICF guidelines developed by the H2020 funded i-consent project. It means to provide information for the development of informed consent on research involving humans | 2021 | No | Guideline |
| Regulatory and Ethics Toolkit, ICF guidelines | GA4GH and IRDiRC have developed model consent clauses for rare diseases research, in order to improve data interoperability, to meet the informational needs of participants, and to ensure proper ethical and legal use of data sources and participants’ overall protection | 2021 | Yes | Guideline |
| Neonatal trials and informed consent: Points to consider | PedCRIN tool: This tool provides a checklist of practical points to consider when talking to parents about the possible inclusion of a neonate into a clinical trial | 2021 | Yes | Recommendation |
| Recruitment and informed consent procedure template | Template for describing recruitment arrangements and/or informed consent procedure for clinical trials within the scope of the Clinical Trial Regulation (CTR). This template has been endorsed by the EU Clinical Trials Coordination and Advisory Group (CTAG) to comply with CTR | 2022 | No | Guideline |
| Effective eConsent Strategies for Every Study: Utilizing the eConsent Fit-for-Purpose Study Framework | This tool provides sponsors (and any stakeholder interested in eConsent) a guide on how to define and design the right eConsent for a particular study and how to generate effective and comparable study data on eConsent. | 2024 | No | Recommendation |
| Guidelines for Good Operational Practice Version 3.0 | Swiss Clinical Trial Organisation Tools: The Guidelines for Good Operational Practice (GGOP) are a framework of common standards for professional and operational practice in clinical research. | 2017 | No | Guideline |
| Easy Guide to Clinical Studies (Easy GCS) Beta | Swiss Clinical Trial Organisation tool: This interactive guide provides comprehensive and concise information and guidance on how to plan and conduct your study. | 2023 | No | Guideline |
| Recommendations on cross-border access to clinical trials | To address the current challenges faced by cross-border participants in clinical trials, the EU-X-CT initiative has developed general recommendations on key aspects of clinical trial management and conduct. The recommendations aim to improve safe and equitable access to cross-border clinical trials within Europe. | 2025 | No | Recommendation |
| Trials@Home recommendations on the conduct of decentralised clinical trials | The Trials@Home consortium, a public-private partnership funded by the Innovative Medicines Initiative has published a comprehensive set of recommendations covering methodological, regulatory, ethical, technical, operational, and social aspects of Decentralized Clinical Trials. | 2025 | No | Recommendation |
| Recommendation paper on decentralized elements in clinical trials | Recommendations on the introduction of decentralised elements in the conduct of clinical trials in the EU/EEA, regardless of any health crisis, and in consideration of the currently limited national guidances. | 2025 | No | Recommendation |
| ERDERA Management of multinational clinical trials for rare diseases | This three‑hour programme familiarises investigators and site teams with the specific challenges of initiating and managing multinational rare‑disease trials. It is solution‑oriented, addressing common barriers such as differing national implementations of European clinical research Regulations and ethical requirements. | 2025 | Yes | Training |
REGULATORY SUBMISSION
Prior to initiating a clinical trial, researchers must obtain approval from National Competent Authorities (NCA) and ethics committees. For clinical trials on an Investigational Medicinal Product (CTIMP), there are a number of steps to follow:
- Compliance with legislation
The first step is to confirm, for international clinical trials in Europe, whether it falls within the scope of European Legislation (CTR-Clinical trials Regulation EU No 536/2014).
The Regulation has introduced an authorisation procedure based on a single submission via a single EU portal (CTIS), an assessment procedure leading to a single decision, rules on the protection of subjects and informed consent, and transparency requirements.
Non-international trials (single-country) would follow national legislation with a single application to the competent authority in the participating country.
- Submission to National Competent Authorities and Ethics Committees
To obtain authorization for clinical trials under the CTR, clinical trials have to be submitted through the Clinical Trials Information System (CTIS) — the centralized portal established by the European Medicines Agency (EMA) . Clinical Trial Applications (CTAs) are based on the submission of a dossier including:
- Part I: General trial information (design, IMP, safety, scientific data) — evaluated jointly by all Member States Concerned (MSCs)
- Part II: National-specific information (ethics, informed consent, compensation) — evaluated individually by each MSC
MSCs review the application within regulated timelines and once approved, the trial is authorized in participating countries. Summary information is automatically published on the EU Clinical Trials Register (public CTIS interface). After registration, CTIS is also used to:
- Submit substantial modifications
- Report serious breaches, safety events, and annual safety reports (ASRs)
- Upload summary results after trial completion
| Tool Name | Relevance | Year | RD/ Paediatric specific | Type |
| EMA list of national competent authorities in the EU | Updated list of European national competent authorities and their contact details | Regular updates | No | Inventory |
| EUREC | List of European Research Ethics Committees in Europe | Regular updates | No | Inventory |
| Regulatory and Ethical Database (RED) | A search tool for regulatory requirements on clinical trials per country. Launched in December 2015 by the European Clinical Research Infrastructure Network (ECRIN) – this is an online database including country-specific information on regulatory and ethical requirements in clinical research across Europe | 2015 | No | Search tool |
| Comprehensive Inventory STARS | The STARS (Strengthening Regulatory Science) project has developed an online Comprehensive Inventory that assists European academic drug developers in finding various support services provided by NCAs, public actors and private entities. The inventory lists various support services including assistance in clinical trial applications | Regular updates | No | Inventory |
| REGULATION (EU) No 536/2014 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 16 April 2014 on clinical trials on medicinal products for human use | The latest EU regulation for clinical trials on medicinal products for human use replaces national laws and describes the rules for assessing clinical trial applications and conducting clinical trials throughout the EU | 2015 | No | Legislation |
| International Compilation of Human Research Standards, 2021 Edition | US Department of Health and Human Services developed listing of over 1,000 standards on human research standards in 131 countries and from many international organizations. These standards may include laws, regulations, and/or guidelines | 2024 | No | Inventory |
| Clinical Trials Regulation (EU) No 536/2014 in Practice | Quick guide on the rules and procedures of the EU Clinical Trials Regulation drawn up by the Clinical Trials Coordination and Advisory Group (CTAG) as its members are the National Contact Points defined in the abovementioned Regulation. | Regular updates | No | Guideline |
| Eudralex - Templates CTIS Application Part I and Part II CTA | Templates documents for Part I and Part II documentation for CTIS applications | Regular updates | No | Guideline |
| MedTechEthics, National Part II CTA Requirements | This report provides an overview of the required part II documentation of 21 MS in EU/EEA. | Regular updates | No | Guideline |
QUALITY MANAGEMENT
The sponsor should implement a system to manage quality throughout all stages of the trial process, in particularly on trial activities essential to ensuring human subject protection and the reliability of trial results.
Quality management is the overall process of establishing and ensuring the quality of processes, data, and documentation associated with clinical research activities. It encompasses both quality control (QC), and quality assurance (QA) activities.
- Quality control. The operational techniques and activities undertaken within the quality assurance system to verify that the requirements for quality of the trial-related activities have been fulfilled. Periodic operational checks to verify that clinical data are generated, collected, handled, analysed, and reported according to protocol, SOPs (Standard Operation Processes), and GCP (Good Clinical Practice
- Quality assurance. ICH Good Clinical Practice guideline defines quality assurance as all those planned and systematic actions that are established to ensure that the trial is performed and the data are generated, documented, and recorded in compliance with GCP and applicable regulatory requirements.
A quality management system (QMS) provides a framework for all quality management activities, including quality control, quality assurance, quality improvement and the reporting of these activities. Key elements of the QMS are:
- Documented procedures developed, implemented and kept up-to-date
- A documentation system that allows for the retrieval of any records or documentation to show actions taken, decisions made and results. All approved documents and records are version-controlled
- Defined organisational and accountability charts, roles and responsibilities
- Appropriate documented training of personnel to meet the defined competencies of their role, and familiarisation with GCP
- Documented evidence to demonstrate that computerised systems are fit for purpose (validation)
- Quality Control (QC) activities, (review and checking). For example, monitoring of trial sites either on-site or through centralised or remote monitoring techniques
- Quality Assurance (QA), including independent audit of QMA processes and studies by the QA team
- A risk-based approach used to determine the extent of trial monitoring activities, processes and trials to audit, and computer validation activities
- Continuous improvement incorporating Corrective and Preventive Actions (CAPA)
Monitoring and Audit
Many professionals working in clinical research may not appreciate or understand the roles and differences between clinical research monitoring and auditing, two distinctly different functions.
Monitoring is a quality control function where study conduct is routinely assessed on an on-going basis at every step of the trial.
Auditing, a quality assurance function, is an independent, top-down, systematic evaluation of trial processes and quality control.
Quality control: Monitoring of clinical sites
ICH- GCP defines monitoring as the act of overseeing the conduct of a clinical trial, that is ensuring that the trial is conducted according to protocol, GCP, SOPs and regulatory requirements. It is the responsibility of the sponsor to ensure the trial is adequately monitored.
According to GCP guidelines, “Monitoring may include site monitoring (performed on-site and/or remotely) and centralised monitoring, depending on the monitoring strategy and the design of the clinical trial (ICH GCP E6 (R3) 3.11.4)The rationale for the chosen monitoring strategy is documented in the monitoring plan.
The monitoring plan sets out monitoring strategies, the monitoring responsibilities of all parties involved, the various monitoring methods to be used, and the rationale for their use. It also describes monitoring procedures, types of visits, what is involved in the conduct of those visits, and the quantity or percentage of each type of document to be monitored. These procedures can be further defined on a protocol basis depending on the purpose, design, size, complexity, and primary outcome measures of the trial.
In general, on-site monitoring is required and remote monitoring may occur at any given research site before a trial begins, while it is in progress, and after it concludes or is terminated. In many instances, study monitors may visit each site after the first one to two participants are enrolled and then schedule subsequent visits based on multivariate criteria, such as the rate of enrolment, volume of data to review, site performance, and other considerations. Study monitors conduct site visits according to the procedures described in the monitoring plan
Regulatory authorities and changes to guideline ICH E6 (R3) recognized the potential use of a risk-based monitoring approach to improve the conduct of clinical trials of all phases
A well-implemented risk-based monitoring process facilitate efficient and cost effective trial delivery without compromising patient safety or data quality.
Documentation of all aspects of the monitoring process is achieved through monitoring reports from on-site visits providing clear evidence of what was checked and any non-compliance as well as the description of associated actions/resolution. Monitoring activities conducted centrally would be required to provide similar evidence. This also enables auditors/inspectors to reconstruct how a trial was managed.
Quality assurance: audits
Audit is defined by ICH as “a systematic and independent examination of trial-related activities and documents to determine whether the evaluated trial-related activities were conducted, and the data were recorded, analyzed, and accurately reported according to the protocol, sponsor’s SOPs, GCP and the applicable regulatory requirement(s)”
Audits are considered good practice and should be part of the Sponsor´s Quality Management system. There are three general areas of concern in the audit process regardless of which entity is performing the audit: patient safety, including consent forms and ethics committees activities; data collection and records; and the pharmacy and investigational drug supply.
Inspections
The ICH defines “inspection” as “The act by a regulatory authority (ies) of conducting an official review of documents, facilities, records, and any other resources that are deemed by the authority (ies) to be related to the clinical trial and that may be located at the site of the trial, at the sponsor’s and/or contract research organization’s (CRO’s) facilities, or at other establishments deemed appropriate by the regulatory authority (ies)”.
Inspections are performed by government regulators to ensure patient safety, welfare, scientific integrity and compliance with regulations. In Europe, regulatory authorities perform regulatory inspections of trial sites or sponsors on a regular basis. The frequency of these inspections is based on the risk associated with the trials being undertaken. They may also perform triggered inspection after a particular event. In most cases the regulatory authority shall inform the main contact (normally sponsor) of an inspection but these may occasionally be unannounced.
| Tool Name | Relevance | Year | RD/ Paediatric specific | Type |
| ICH Good Clinical Practice E6 (R3) | This document addresses the good clinical practice, an international ethical and scientific quality standard for designing, conducting, recording and reporting trials that involve the participation of human subjects. It aims to provide a unified standard for the ICH regions to facilitate the mutual acceptance of clinical data by the regulatory authorities in these jurisdictions | 2024 | No | Guideline |
| ICH Good Clinical Practice E6(R3) Training course (Global Health Network) | This short course aims to provide the researcher with the basic principles of GCP and how these principles can be applied practically in the research setting. The course is aimed at all those involved in clinical research. | 2025 | No | Training |
| ICH Good Clinical Practice E6(R3) Training material (EMA) | EMA has developed Interactive training materials on the Revised ICH E6(R3) principles and Annex 1 | 2025 | No | Training |
| WHO Guidance for best practice for clinical trials | The WHO guidance for best practice for clinical trials provides a comprehensive framework for the design, conduct, registration, oversight, and reporting of clinical trials globally. It aims to ensure ethical, scientific, and methodological integrity in clinical research. | 2024 | No | Guideline |
| EC, Risk proportionate approaches in clinical trials | Recommendations of the expert group on clinical trials for the implementation of Regulation No 536/2014 (EU) No 536/2014 on clinical trials on medicinal products for human use, as per risk based quality management | 2017 | No | Recommendation |
| ECRIN_Risk-Based Monitoring Toolbox | Provides information on tools available for risk assessment, monitoring and study conduct, the institutions where they are used, and other relevant details such as links and user feedback | 2015 | No | Toolbox |
| Guidelines for Risk-Based Monitoring, Version 3.0 | Swiss Clinical Trial Organisation tool: Guidelines for Risk-Based Monitoring | 2022 | No | Guideline |
SAFETY REPORTING
The sponsor is responsible for the ongoing safety evaluation of the Investigational Medicinal Product(s) used in a Clinical Trial
Sponsors should develop formal, written processes (Safety Management Plan) for the management of adverse events and safety reports, including the handling of both expedited reports and annual safety reporting.
Investigators are responsible for Serious Adverse Events (SAEs) reporting to the Sponsor. SAE reporting includes investigator´s causality and seriousness assessment and it must be done timely according to the protocol specifications.
Pharmacovigilance (PV) is the science relating to the detection, assessment, understanding and prevention of the adverse effects of medicines/vaccines. Some academic Clinical Trial Units (CTUs) and private Clinical Research Organizations (CROs) have PV units to support Sponsors and investigators with Safety reporting, including:
- Validation, use and maintenance of an internal PV database in which all the events can be received, managed, and recorded and where all obligatory legal PV activities can be performed in compliance with the national and global regulatory requirements.
- Expedited reports
All relevant Suspected Unexpected Serious Adverse Reaction (SUSARs) that occur during a clinical trial must be reported to the Regulatory authorities of participating countries as soon as possible but no later than 7 calendar days after first knowledge by the sponsor. The communication has to be followed by as complete as possible a report within 8 additional calendar days. Sponsors may use the current European portal (Eudravigilance website) to submit SUSAR reports in bulk to National Competent Authorities (NCAs).
Annual safety reports
Development Safety Update Reports (DSURs)/Annual Safety Report (ASR) are internationally-harmonized, safety documents covering the safety summary of medicinal products during their development or clinical trial phase. Sponsor is responsible of submitting annual ASR via the Clinical Trials Information System (CTIS)once per year to inform both NCAs and Ethics Committees (ECs) in the countries involved in the clinical trial.
- Periodical reconciliation of adverse events recorded between the Clinical trial database and the PV database
- Pharmacovigilance training for investigators and staff involved in the trials.
| Tool Name | Relevance | Year | RD/ Paediatric specific | Type |
| ICH Topic E2A: Clinical Safety Data Management | Notes for definitions and standards on Safety reporting for ICH topic E A 2 | 1995 | No | Guideline |
| ICH Topic E2F Development Safety Update Report | Guidance on Safety Reporting. The Development Safety Update Report (DSUR) proposed in this guideline is intended to be the common standard for annual clinical trial safety reporting among the ICH regions | 2010 | No | Guideline |
| Guideline on good pharmacovigilance practices (GVP) Product- or Population-Specific Considerations IV: Paediatric population | Guidance on the conduct of pharmacovigilance for medicines used by paediatric population. It is directed towards marketing authorisation holders and competent authorities. It is also of relevance to all those involved in the conduct of paediatric clinical trials. | 2017 | Yes | Guideline |
| Safety training | Swiss Clinical Trial Organisation Tools: Safety training to consolidate investigator's knowledge of patient safety and reporting issues in clinical research | Regular updates | No | Training |
| Introduction to collecting and reporting adverse events | This short course provides a general introduction and overview of Adverse Events and how to deal with them when they occur. This course is suitable for everyone involved in clinical research. | Regular updates | No | Training |
| Safety reporting forms | Swiss Clinical Trial Organisation Tools: Set of comprehensive forms for safety reporting tailored to different types of clinical research projects. | Regular updates | No | Template |
| Causality Assessment of Adverse Events in paediatric trials | PedCRIN tool: A visual algorithm based on the Naranjo scale and specifically adapted for the paediatric population to help researchers in their assessment of causality of adverse events occurring during a clinical study | 2021 | Yes | Recommendation |
| Safety data analyses of neonatal trials: Points to consider | PedCRIN tool: This tool provides practical points to consider when planning for the analysis of neonatal safety data | 2021 | Yes | Recommendation |
DATA MANAGEMENT
A process that begins with conception and design of the clinical trial, continues through data capture and analysis to publication, data archiving and data sharing with the broader scientific community. The Data Management Plan (DMP) describes the procedures for data collection and management throughout the lifecycle of a clinical trial.
| Tool Name | Relevance | Year | RD/ Paediatric specific | Type |
| Research Data Management | A guide to managing outputs of research projects and handling issues such as copyright and data protection laws | Regular updates | No | Guideline |
| Research Data Management Kit | This is a web-based resource for research data management. It has been designed to guide life scientists in their efforts to better manage their research data following the FAIR Principles as well as help researchers be more productive for themselves and their collaborators. | 2021 | No | Toolbox |
| Guideline on computerised systems and electronic data in clinical trials | This guideline addresses the use of computerised systems—including instruments, software, and 'as a service' platforms—in the creation, capture, and management of electronic clinical data. It aims to ensure the quality and reliability of trial data, safeguarding participant rights and supporting robust decision-making processes. The guideline applies to systems utilized throughout the clinical trial lifecycle, from planning to archiving. | 2023 | No | Guidelines |
| Data Certification Standards/Data Certified Units | The ECRIN Data Centre Certification programme identifies non-commercial clinical trials units (CTUs) in Europe that have demonstrated they can provide safe, secure, compliant and efficient management of clinical research data | 2018 | No | Recommendation |
| EDCTP Data Management portal | An initiative of the Global Health Network. This tool helps to identify the areas to consider when developing a Data Management Plan, with a particular focus on data management systems and how to organise and structure data. Includes best practices for data capture, entry, processing and monitoring and how to prepare your data for analysis, sharing and archiving. | Regular updates | No | Toolbox |
| EDCTP Data Sharing Toolkit | An initiative of the Global Health Network, this Data Sharing Toolkit, collates practical information and resources related to data sharing, including data management basics, data sharing steps and a repository finder | Regular updates | No | Toolbox |
| Sharing and reuse of individual participant data from clinical trials: principles and recommendations (Ohmann et al., 2017) | This article lists recommendations on providing access to individual participant data from clinical trials, using a broad interdisciplinary approach | 2017 | No | Article |
| Evaluation of repositories for sharing individual-participant data from clinical studies (Banzi et al., 2019) | This article analyses the current landscape of data repositories to create a detailed description of available repositories and assess their suitability for hosting data from clinical studies, from the perspective of the clinical researcher | 2019 | No | Article |
| Mapping of Data-Sharing Repositories for Paediatric Clinical Research—A Rapid Review (Felisi et. al., 2024 | This paper looked at existing data-sharing repositories (DSRs) for the re-use of individual paediatric patient data from clinical trials | 2024 | Yes | Article |
| Sharing and reuse of health-related data for research purposes: WHO policy and implementation guidance | This document clarify the policy and practice on the reuse and onward sharing for research purposes of health data collected under the auspices of WHO technical programmes. It covers use in both emergency and non-emergency situations. | 2022 | No | Guideline |
| Guidelines 01/2025 on Pseudonymisation European Data Protection Board | These guidelines intend to clarify the use and benefits of pseudonymisation for controllers and processors | 2025 | No | Guideline |
| Guidelines for data sharing of investigator-initiated clinical studies | This document provide general guidance for preparing data sharing plan for clinical studies, including guidance on informed consent (ICF) for secondary use of data and long-term storage of data, especially Individual Patient Data (IPD) in repositories following the FAIR (Findable, Accessible, Interoperable and Reusable) principles. | 2024 | No | Recommendation |
INVESTIGATIONAL PRODUCT/SUPPLY MANAGEMENT
An investigational product (IP), as defined by the ICH is a pharmaceutical form of an active ingredient or placebo being tested or used as a reference in a clinical trial, including a product with a marketing authorization when used or assembled (formulated or packaged) in a way different from the approved form, or when used for an unapproved indication, or when used to gain further information about an approved use.
Regulation (EU) No 536/2014 Article 2 (5) defines an “Investigational Medicinal Product (IMP) as “a medicinal product which is being tested or used as a reference, including as a placebo, in a clinical trial”.
To ensure all regulatory and governance requirements are met, it is essential that investigators obtain advice and support from those with specialist knowledge relating to the IMP supplies (Clinical Trial Pharmacists, Clinical Trials Unit (CTU) or Contract Research Organisation (CRO)). These specialists are key to plan and advice on IMP handling. In Europe, IMP handling requires a thorough knowledge of ICH GCP sections on Supply and handling IMPs and Eudralex volumes on Good Manufacturing Practice (GMP).
It is highly recommended to start considering IMP handling from the planning stage of the trial, clearly identify the nature of the IMP and a thorough consideration of the IMP sourcing strategy. The complexity of the process varies greatly from IMPs sourced internally and dispensed according to routine practice to externally sourced IMPs not marketed. The IMP requirements relating to unlicensed drugs also applies to compounds that already marketed, for example when an IMP is used for a new indication outside those for which the drug was licensed, such as for a different group of patients or for a different disease. This is especially relevant on the rare diseases clinical research, where a good number of investigator-initiated trials aim to generate knowledge on drug repurposing.
| Tool Name | Relevance | Year | RD/ Paediatric specific | Type |
| EudraLex - Volume 10 - Clinical trials guidelines_Chapter III_Quality of the investigational medicinal product. | Volume 10 of the publication "The rules governing medicinal products in the European Union" contains guidance documents applying to clinical trials. | Regular updates | No | Guideline |
| EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use | Good manufacturing practice (GMP) describes the minimum standard that a medicines manufacturer must meet in their production processes. The European Medicines Agency (EMA) coordinates inspections to verify compliance with these standards and plays a key role in harmonising GMP activities at European Union (EU) level. The Annex also includes guidance on ordering, shipping, and returning clinical supplies, which are at the interface with, and complementary to, guidelines on Good Clinical Practice. | Regular updates | No | Guideline |
| Guideline on pharmaceutical development of medicines for paediatric use | The principles of this guideline should be considered during the pharmaceutical development of all paediatric medicines as proposed in marketing-authorisation applications (MAAs) or applications to extend or vary marketing authorisations to the paediatric population (MAVs) | 2013 | Yes | Guideline |
| Clinical trials toolkit: Trial Supply | Guide prepared by MODEPHARMA to codify good practice on drug management in publicly funded clinical trials | 2018 | No | Toolbox |
LABORATORY PROCESSES
The analysis of samples collected from subjects participating in clinical trials forms a key part of the clinical trials process. Sample analysis or evaluation provides important data on a range of endpoints which is used, for example, to assess the pharmacokinetic profile of investigational medicinal products and to monitor their safety and efficacy. Consequently, it is essential that sample analysis or evaluation is performed to an acceptable standard which will ensure patient safety is not compromised and that data is reliable and accurately reported.
It is good practice to have well documented Standard Operating Procedures (SOPs) that define how to conduct each laboratory procedure, whether at the clinic or laboratory (analytical) level from the sampling to the analysis and further storage or destruction as applicable.
These applies to:
- samples collected and analysed as part of routine clinical care, that may also contribute to the study dataset
- samples collected and analysed for study objectives only
- samples prepared and stored before shipping to a specialist laboratory
- samples analysed using a standard clinical assay in laboratory with a functional, audited quality system
- samples analysed using an exploratory/experimental assay
- samples collected for biobanking
| Tool Name | Relevance | Year | RD/ Paediatric specific | Type |
| Collection, storage and use of biological samples and related data in paediatric trials | PedCRIN tool: A checklist developed to help researchers, sponsors, and other affiliated personnel verify that all key aspects required to properly manage samples and related data in the context of paediatric trials are taken into consideration | 2021 | Yes | Checklist |
| Reflection paper on laboratory processes for clinical trials | The purpose of this reflection paper is to provide laboratories that perform the analysis or evaluation of human samples collected as part of a clinical trial, with information that will help them develop and maintain quality systems which will comply with relevant European Union Directives, national regulations and associated guidance documents. It will also provide information on the expectations of the inspectors who may be assigned by national monitoring authorities to inspect facilities that perform work in support of human clinical trials | 2010 | No | Reflection paper |
| GCLP (Good clinical laboratory practice) Guidance | This guidance identifies systems required and procedures to be followed within an organization conducting analysis of samples from clinical trials in compliance with the requirements of Good Clinical Practice (GCP). It thus provides sponsors, laboratory management, project managers, clinical research associates (CRAs) and quality assurance personnel with the framework for a quality system in analysis of clinical trial samples, ensuring GCP compliance overall of processes and results. | 2009 | No | Guideline |
| GCP Lab guidance | MHRA Guidance on the maintenance of regulatory compliance in laboratories that perform the analysis or evaluation of clinical trial samples | 2009 | No | Guideline |
| Good clinical laboratory practice training | Good Clinical Laboratory Practice (GCLP) guidelines describe the application of those Good Laboratory Practice principles that are relevant to the analyses of samples from clinical trials while ensuring the purpose and objectives of the Good Clinical Practice principles are maintained. | Regular updates | No | Training |
STATISTICAL ANALYSIS PLAN (SAP)
The SAP is intended to be a comprehensive document that contains a detailed and technical description of the principal features of the statistical analysis outlined in the protocol including detailed procedures for executing the statistical analysis of the primary and secondary endpoints and other data. As compared to the protocol, the SAP should contain an in-depth description of the statistical methods to be used and a definition of the statistical output which will be included in the clinical study report.
The SAP is generally developed as a separate document and written after the protocol has been finalised. Ideally, a biostatistician should develop the SAP with the help of the principal investigator and in alignment with the protocol. The SAP should be finalised prior to data analysis and before treatment un-blinding. Any changes between the methods in the protocol and analysis plan, should be explained in the SAP.
The SAP must properly explain the statistical analysis following the aims and primary objectives, secondary objective, exploratory objectives, primary/secondary/exploratory endpoints, trial population, design of the trial, sample size calculations with justifications/assumptions and the randomization methods. Additionally, a SAP must describe in detail the statistical methodology i.e. efficacy analysis, safety data analysis, interim analyses, handling of missing data, reporting conventions, etc
| Tool Name | Relevance | Year | RD/ Paediatric specific | Type |
| ICH Statistical Principles for clinical trials E9 | This guidance is intended to give direction to sponsors in the design, conduct, analysis, and evaluation of clinical trials of an investigational product in the context of its overall clinical development. The document will also assist scientific experts charged with preparing application summaries or assessing evidence of efficacy and safety, principally from clinical trials in later phases of development. | 1998 | No | Guideline |
| ICH E9 (R1) addendum on estimands and sensitivity analysis in clinical trials to the guideline on statistical principles for clinical trials | The addendum aims to improve the alignment between clinical trial objectives and statistical analyses by introducing the concept of "estimands" — precise descriptions of the treatment effect a trial intends to estimate. This approach facilitates clearer communication among clinicians, statisticians, and regulators. | 2020 | No | Guideline |
| Guideline for the Content of Statistical Analysis Plans in Clinical Trials | A checklist of 32 minimum items for inclusion in SAPs that was developed with the primary intention of being applicable to the final analyses of later-phase randomized clinical trials addressing the minimum recommended content of a SAP | 2017 | No | Guideline |
| ECRIN Metadata Repository | Search of metadata (including protocols and Statistical Analysis Plans) on published clinical trials. | 2020 | No | Repository |
| Clinical Trials Registry (NIH) | Database of publicly and privately funded clinical studies conducted around the world. The Study Documents tab allows search for SAP (Statistical Analysis Plan), protocols and Informed consents | Regular updates | No | Registry |
ARCHIVING
The documents which individually and collectively permit evaluation of the conduct of a clinical trial and the quality of the data produced are defined as essential documents according to the ICH Good Clinical Practice.
These documents service to demonstrate the compliance of the investigator, sponsor and trial monitors with the standards of GCP and with applicable regulatory requirements. It is the responsibility of the sponsor to ensure that the documents are filed in an organised way in the so-called Trial Master File (TMF) which will facilitate management, audit and inspection of the clinical trial.
Essential documents must be retained (archived) for sufficient periods to allow for audit and inspection by regulatory authorities and should be readily available upon request.
The TMF should be set up at the beginning of a trial and maintained throughout the trial. Archiving applies to both the investigator sites and the central trial coordinating office.
Archival Strategy/Archiving plan
Prior to initiation of a trial, an archiving strategy must be developed, which addresses the regulatory requirements. It should include the following elements:
Documents/material to be archived
The plan/strategy must identify the documents, data, supplies, specimens, etc. that are to be archived. Some examples are: source documents, case record forms, informed consent documents, investigator site file, biological samples (if applicable), trial supplies (if applicable), etc.
Destruction of documents
The reasons for destruction of essential documents should be documented and signed by a person with appropriate authority. The sponsor/someone on behalf of the sponsor should notify investigators in writing when their trial records can be destroyed
Period of archiving
The plan must indicate for how long the documents will be archived in compliance with the regulatory guidelines. It is also important that access to documents and data is maintained throughout the storage period. This includes ensuring system maintenance (hardware and software) to access data in the original archived format, or the use of a new system to emulate old software, or the migration of data into a new format to ensure continuous access with new software. This issue should be addressed through written procedures by the organisation responsible for long-term archiving
According to the European Medicines Agency (EMA) the long-term archiving of the TMF can be either through an external archive that provides the preservation of paper documents or through an electronic archiving (e.g., cloud data center). When an external repository is used, the sponsor must still make an assessment of the suitability of the solution used both before use and periodically during the long-term archiving period.
In Europe, according to the Regulation EU No 536/2014 (CTR)sponsor and investigator must archive the content of the clinical Trial Master File (TMF) and Investigator Site File (ISF) for at least 25 years after the end of the clinical trial. However, the subject’s medical records at the trial site shall be archived in accordance with national law. Indeed, the CTR leaves the exact retention period for medical records to each Member State’s legislation, which often exceeds 15 years (and may be linked to hospital or data protection laws).
Location and access
Essential records should be maintained in a legible condition and prompt retrieval should be possible. Adequate and suitable space should be provided for the secure storage of all essential records upon trial completion. The facilities should be secure, with appropriate environmental controls and adequate protection from fire, flood and unauthorized access. The storage of the sponsor's documentation may be transferred to a sub-contractor (e.g. a commercial archive) but the ultimate responsibility for the quality, integrity, confidentiality and retrievability of the documents resides with the sponsor.
The plan must specify that the documents will have a restricted access. It is advisable to identify the details of personnel that can access the archival facility. However, it should be clarified that the regulatory authority or ethics committees may request access to these documents.
The archiving plan should include a disaster management plan, to recover the documents in case of an emergency. For example, electronic copies can be made of all documents and data that are archived. However, these electronic copies must be stored at a location other than the archival facility and comply with confidentiality requirements.
Procedure
It should be specified that the process of archiving should only be commenced after:
- The final visit has been completed by the last trial participant
- TMF and ISF, are complete
- The data has been verified for accuracy
- All queries pertaining to data have been resolved, database is locked, and the data has been analysed
- Final report has been submitted
- Any other pre-requisite specified by the sponsor, regulatory authority, ethics committee, etc.
| Tool Name | Relevance | Year | RD/ Paediatric specific | Type |
| Guideline on the content, management and archiving of the clinical trial master file (paper and/or electronic) | The EMA guideline on the content, management and archiving of the TMF provides guidance relating to the media used for storage of documents (including requirements when original records are transferred to electronic media for the purpose of archive). | 2018 | No | Guideline |
TRIAL REPORT
A Clinical Study Report (CSR) is a is a key document that describes the methodology and results of a clinical trial in drug development.
The full CSR represents a comprehensive clinical and statistical description of a sponsor’s study conduct. Additionally, a full CSR includes efficacy and safety data. This report is required if the study is to be used to support approval by a regulatory agency, such as the FDA (Food and Drug Administration-USA) or European Medicines Agency (EMA), or that support the information in the product label.
A CRS should also provide enough individual patient data, to allow the key analyses of data to be repeated, should the regulatory authorities wish to do so.
In the European Union, sponsors must submit the CSR summary within one year of the end of their clinical trial to regulatory authorities and ethics committees through the Clinical Trials Information System (CTIS). The end of a clinical trial is usually defined as the date of the last patient last visit (LPLV). The results of most clinical trials are published in the EU clinical trials registry and are therefore accessible to the general public. For trials exclusively involving minors, the CSR deadline is 6 months after the end of the trial. This accelerates transparency for paediatric research.If a CSR is not required for regulatory submission (e.g., exploratory early-phase trials), the sponsor may submit only the summary results within the same 12-month window.
The Clinical Trials Regulation (CTR) allows deferral of CSR publication (not submission) in certain cases:
- Sponsors can request deferral of the public disclosure of CSR or summary results in CTIS to protect commercially confidential information.
- However, the CSR itself must still be submitted to CTIS within the 12-month (or 6-month) timeline.
Standard components of an ICH-compliant CSR include:
- Title Page and Synopsis
- Table of Contents, List of Tables/Figures
- Ethics and Administrative Structure
- Introduction
- Study Objectives
- Investigational Plan (Protocol Overview)
- Study Population and Methods
- Efficacy Evaluation
- Safety Evaluation
- Discussion and Overall Conclusions
- References
- Appendices
- Protocol and amendments
- Sample CRFs
- Investigator CVs
- Audit certificates
- Individual patient data listings
| Tool Name | Relevance | Year | RD/ Paediatric specific | Type |
| TranspariMED: Clinical trial transparency tools | Collection of hands-on tools and case studies that universities and other institution can use to improve their registration and reporting of clinical trials. | 2019 | No | Toolbox |
| ICH E3 guideline on Structure and Content of Clinical Study Reports (ICH E3) | The objective of this guideline is to allow the compilation of a single core clinical study report (CSR) acceptable to all regulatory authorities of the ICH regions | 1995 | No | Guideline |
| Consort 2025 statement | CONSORT Statement is an evidence-based, minimum set of recommendations for reporting randomized trials. It offers a standard way for authors to prepare reports of trial findings | 2025 | No | Checklist |
| Guidelines for Reporting Outcomes in Trial Reports: The CONSORT-Outcomes 2022 Extension | The CONSORT-Outcomes 2022 extension provides evidence- and consensus-based guidance for reporting outcomes in published clinical trial reports, extending the CONSORT 2010 statement checklist with 17 additional reporting items and harmonizing reporting ecommendations with guidance from the SPIRIT-Outcomes 2022 extension. | 2022 | No | Checklist |
| CONSORT statement on N-of-1 trials (extension of the CONSORT Statement) | A CONSORT extension for N-of-1 trials that provides guidance on the reporting of individual and series of N-of-1 trials | 2015 | Yes | Checklist |
DISSEMINATION
After each clinical trial finishes, the trial sponsor will compile a detailed clinical study report (CSR), which follows a format laid down by the regulatory authorities. Access to the complete CSR is usually limited to the sponsor and the regulatory authorities that are assessing the marketing authorisation application.
However, information that has been summarised from the CSR is likely to come into the public domain by different means:
Journal papers-The classic route for publication of clinical trial results is a research paper in a specialist medical journal, by which the results are published as an article subject to a peer-review process. Current practice usually involves the publication of the full study protocol as well.
Conferences-CT Results are usually presented at international medical conferences. Nevertheless, access to this information is often restricted to those who are attending the conference and it is not easily available to those who are not.
Clinical trials registries - In Europe the European Clinical Trials register (https://euclinicaltrials.eu/) allows to search clinical trials initiated in the European Union and European Economic Area since 31 January 2022. For trials taking place in the EU starting after 1st January, 2015, all such results must be published, regardless of their positive or negative implications. The World Health Organisation (WHO), through its International Clinical Trials Registry Platform, is setting international standards for registering and reporting on all clinical trials. In the United States (US), the ClinicalTrials.gov registry does so similarly.
EPAR-When authorisation for a new medicine is sought via the Centralised Procedure (CP), an assessment report (EPAR) is written by the European Medicines Agency (EMA), published on EMA website after a decision has been made either to approve or reject the authorisation application. The EPAR provides public information on a medicine, including how it was assessed by the EMA committees
Lay summary report- The European Union Clinical Trials Regulation (EU CTR) 536/2014 includes a requirement for the submission of lay summaries. As a complement to other forms of clinical study disclosure such as registry postings and scientific publications, lay summaries may aid the transparency of a sponsor’s clinical study results, thereby promoting trust, partnership, and patient engagement throughout the clinical study process.
| Tool Name | Relevance | Year | RD/ Paediatric specific | Type |
| EUPATI tutorial: Reporting and recording clinical trial results | EUPATI tutorial about recording and reporting clinical trial results | Regular updates | No | Training |
| EudraCT (European Union Drug Regulating Authorities Clinical Trials Database) | European Clinical Trial CT Database. Since 2014, it is the responsibility of sponsors to ensure that the protocol information and results of all clinical trials is submitted in EudraCT; this information is publicly available through the EU Clinical Trials Register (EU CTR). | Regular updates | No | Registry |
| Technical guideline on the format of the data fields of results-related information on clinical trials | Technical guideline on the format of the data fields of results-related information on clinical trials to publish on the EU Clinical Trials Register | 2013 | No | Guideline |
| Tutorials on posting results on EudraCT | Tutorials on posting results on the European Union Drug Regulating Authorities Clinical Trials Database (EudraCT) | Regular updates | No | Training |
| Summaries of clinical trial results for laypersons | Guideline provides sponsors and investigators with guidelines and templates for the production of summaries of clinical trial results for laypersons | Regular updates | No | Guideline |
| COBWEB Consort-based WEB tool an online writing aid tool for writing a randomized trial report | COBWEB is an online manuscript writing aid tool intended to guide authors through the process of manuscript writing of Randomized Controlled Trials (RCTs) in line with the Consolidated Standards of Reporting Trials (CONSORT) and its subsequent extensions. | Regular updates | No | Tool |

The toolbox has been developed through funding from the EJP-RD project.
![]() The European Joint Programme on Rare Diseases is an initiative that has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement N°825575
The European Joint Programme on Rare Diseases is an initiative that has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement N°825575